Authors: Marcus Fernando Kodama Pertille Ramos and Ítalo Beltrão Pereira Simões
The majority of gastric cancer (GC) cases are sporadic, but about 10% present familial aggregation and 1 to 3% have a hereditary cause. Knowledge of hereditary syndromes as a causal factor for colorectal cancer (CRC) is well estabilished, but for GC this is less publicized, which can impair early diagnosis and proper follow-up.
Hereditary GC can occur with or without the presence of polyposis, as in the case of Hereditary Diffuse Gastric Cancer, Li-Fraumeni, BRCA1, BRCA2, and Lynch syndromes. In this article, we will briefly describe the main hereditary GC syndromes associated with polyposis, leaving those not associated for a future article.
1. HEREDITARY GC ASSOCIATED WITH POLYPOSIS
1.1 FAMILIAL ADENOMATOUS POLYPOSIS (FAP)
FAP results from a mutation in the APC tumor suppressor gene causing a very high risk of CRC.
About 51 to 88% of patients have gastric polyps, mainly fundic gland polyps (FGP). The incidence is high even in attenuated FAP. They tend to be numerous, and the term gastric polyposis can be used only when more than 20 are present.
Low-grade dysplasia may be present in up to 44% of fundic gland polyps. Adenomatous polyps are detected in about 20% of patients with FAP.
Upper endoscopic screening is recommended at the time of colonic polyposis manifestation or from the age of 25. The interval of performance will depend on the findings and also according to the need for follow-up of papillary adenoma, when present, according to the Spigelman score.
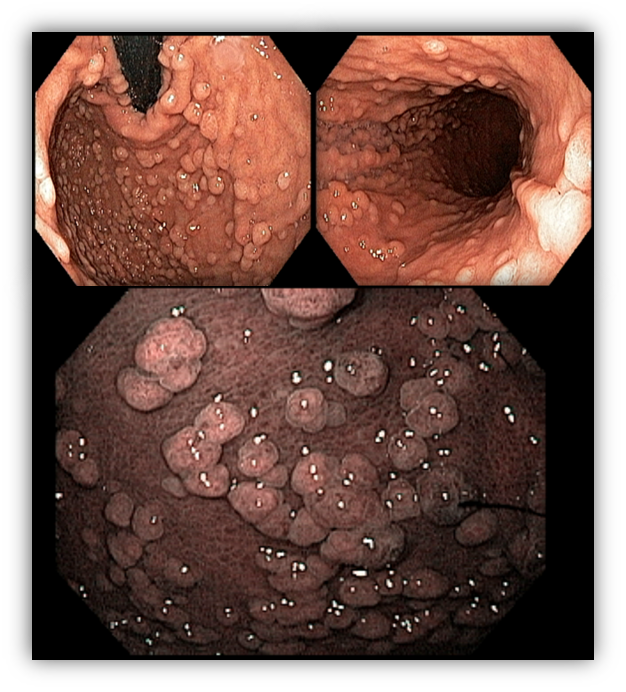
1.2 PEUTZ-JEGHERS SYNDROME (PJS)
PJS is an autosomal dominant disorder, characterized by the development of gastrointestinal hamartomatous polyposis mainly in the jejunum associated with the presence of melanocytic macules.
The clinical diagnosis is based on the confirmation of the presence of hamartomatous polyps associated with a positive family history and hyperpigmentation of mucous membranes, fingers, and external genitalia.
Gastric polyps are detected in 25% of cases, compared with 70-90% found in the small intestine and 50% in the colon. The morphological appearance of the gastric polyp in PJS resembles a villous pattern of hyperplastic epithelial proliferation, making it difficult to distinguish from juvenile and hyperplastic polyps.
Dysplasia is rarely detected in the polyps, but individuals with PJS have a 29% risk of developing GC, mainly of the intestinal type.
Screening should be started early in childhood with initial endoscopy, with frequency depending on the findings. From the age of 50, the risk of GC increases and the frequency should be more frequent between 1 to 2 years.
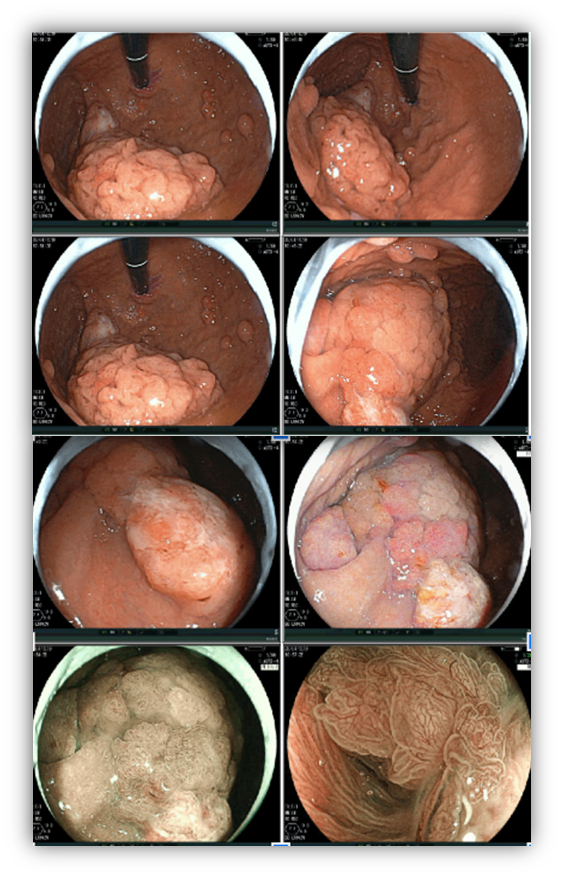
1.3 JUVENILE POLYPOSIS SYNDROME
An autosomal dominant syndrome that leads to the development of polyps throughout the gastrointestinal tract mainly in the colon and rectum.
Criteria for clinical suspicion of the syndrome include more than 5 colorectal juvenile polyps, juvenile polyps throughout the gastrointestinal tract, or more than 1 juvenile polyp with a positive family history. The definitive diagnosis is made from one of the clinical suspicion criteria in the presence of the BMPR1A and SMAD4 genes in the genetic test.
Juvenile polyps are hamartomatous polyps that develop from normal tissue of the gastrointestinal tract. The usual endoscopic appearance is of a pedunculated, multilobed, soft polyp ranging from small polyps to giant polyps. In up to 75% of cases, other types of polyps are present together. Severe gastric polyposis can occur causing anemia, hematemesis, protein-losing enteropathy, and obstructive symptoms. Progression to GC occurs in up to 21% of cases with an average age of 58 years.
Endoscopic screening is recommended from adolescence with annual endoscopies.
Learn more about juvenile polyposis by clicking this post
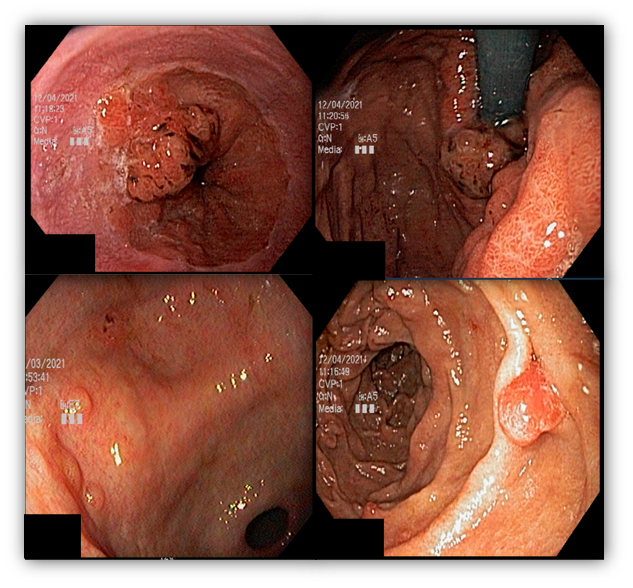
1.4 MUTYH-ASSOCIATED POLYPOSIS (MAP)
MAP is a rare syndrome, autosomal recessive, associated with mutation in the MUTYH gene that participates in DNA repair processes. Patients with MAP are predisposed to CRC, breast, and ovarian cancer.
Gastric polyps are detected in about 10 to 33% of cases and the majority are adenomas and FGP.
The risk of GC is low (2%) but occurs in younger patients (median age of 38 years). On the other hand, the risk of duodenal cancer is high and can occur in 17% of cases.
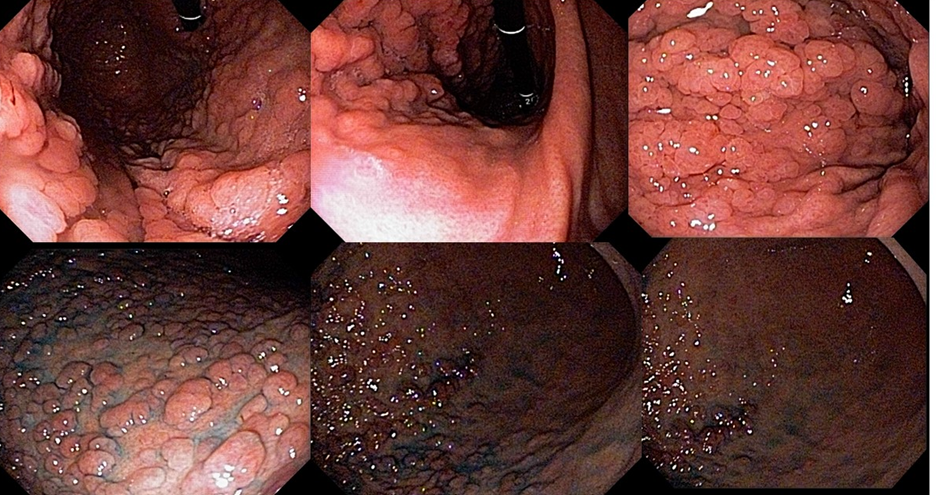
1.5 GASTRIC ADENOCARCINOMA WITH PROXIMAL POLYPOSIS (GAPPS)
This syndrome is characterized by the development of proximal gastric polyposis including the fundus and body forming a carpet of small polyps usually smaller than 1 cm. The histological type of the polyps is varied and can be FGP, hyperplastic, adenomas, and mixed.
The criteria for clinical diagnosis include detection of more than 100 polyps or more than 30 polyps with a positive family history in a first-degree relative, polyps restricted to the body and fundus without the presence of colorectal polyps, morphology of FGP with areas of dysplasia or carcinoma, exclusion of other syndromes, and use of proton pump inhibitors.
Case series have reported an incidence of 12.7% of GC, all of the intestinal type.
Endoscopic follow-up should be performed, but in cases with multiple polyps, the evaluation of polyps with signs of degeneration may be impaired, indicating total gastrectomy.

References
- Clauditz TS, Moore M, Setia N, et al. Syndromic gastric polyposis and hereditary gastric cancers. Diagnostic Histopathologic 2019; 26(1):39-46.
- Mahon SM. Hereditary Polyposis Syndromes. Gentics and Genomics 2018; 22(2): 151-6
- Cardoso DM. Síndromes de polipose colorretal. Endoscopia Terapêutica; 2020. Disponível em: http://endoscopiaterapeutica.com.br/assuntosgerais/sindromes-de-polipose-colorretal/
How to cite this article
Kodama, MFKP and Simoes IBP. Hereditary gastric cancer. Gastropedia 2022. Available at: https://endoscopiaterapeutica.com.br/assuntosgerais/cancer-gastrico-hereditario